Basic
Basic.RmdCount K-mers in FASTQ Files
We are using simulated k-mer data from the phi-X174 genome.
Getting the data and setting parameters:
suppressPackageStartupMessages( library(kmerize) ) fq <- system.file("testdata/phix174-pe_w_err_5k_30q.fastq.gz", package = "kmerize") k <- 9 out_file <- file.path(tempdir(), "phwei11") out_db <- paste0(out_file, c(".kmc_pre", ".kmc_suf"))
Counting:
if (kmerize:::check_install_ok()) { kmer_path = kmr_count(fq, out_file, k = k, f = "q" # file input format is fastq ) }
Convert to readable tabulated database:
if (kmerize:::check_install_ok()) { kp <- kmr_write_tab(kmer_path) kmers <- kmr_read_tab(kp) head(kmers) } #> # A tibble: 6 x 2 #> kmer count #> <chr> <dbl> #> 1 AAAAAAAGT 27 #> 2 AAAAAACGT 37 #> 3 AAAAAAGCC 38 #> 4 AAAAAAGTT 27 #> 5 AAAAAATTT 52 #> 6 AAAAACATT 25
Genomic Response
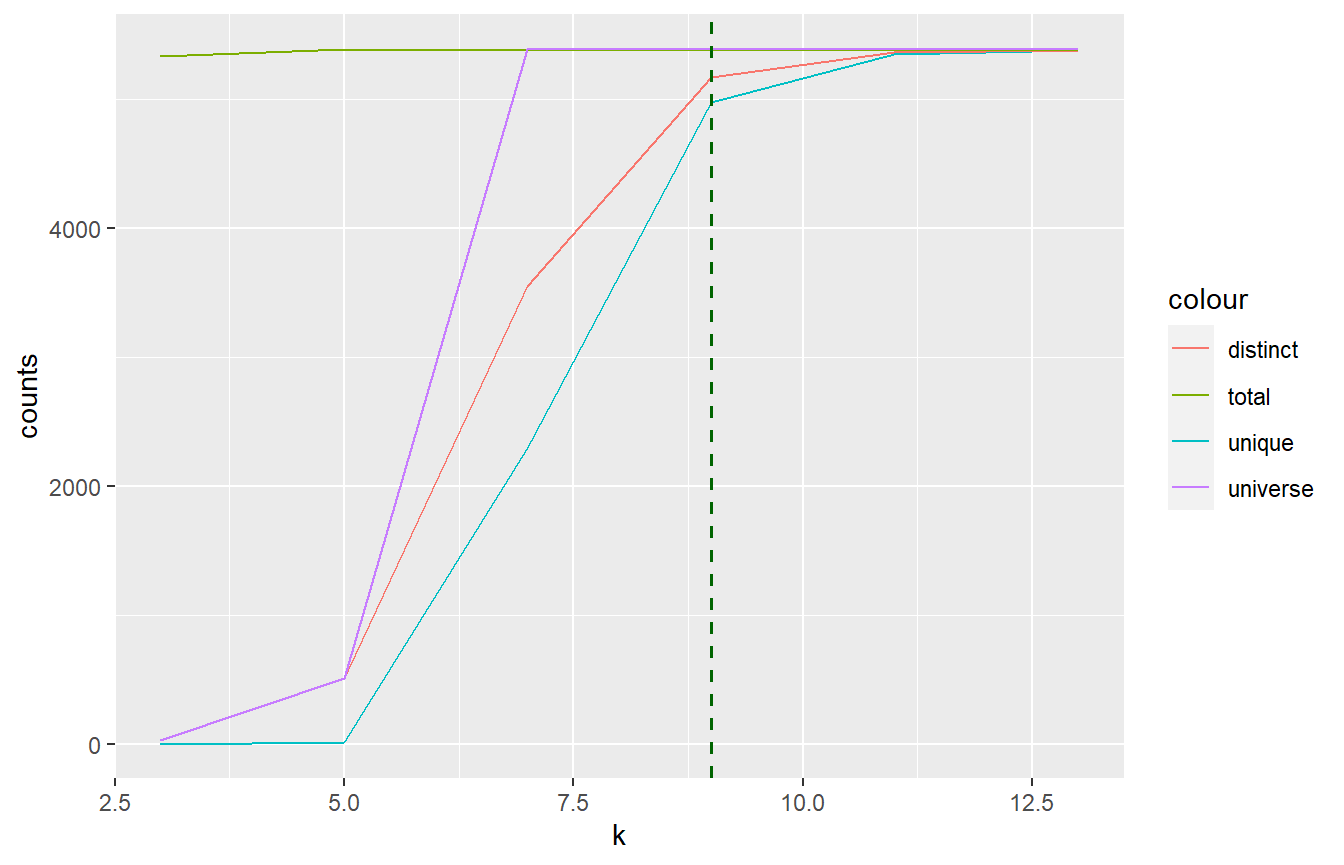
# Getting the DNA if (kmerize:::check_install_ok()) { fp <- system.file("testdata/phix174.fasta", package = "kmerize") dna <- Biostrings::readDNAStringSet(fp) k <- as.integer(seq(3, 25, 2)) # currently k explicitly as integer sequence res <- kmr_response(fp, k, fmt = "m") res } #> ===== #> k unique distinct total universe #> 1 3 0 32 5328 32 #> 2 5 10 506 5382 512 #> 3 7 2295 3547 5380 8192 #> 4 9 4972 5170 5378 131072 #> 5 11 5346 5361 5376 2097152 #> 6 13 5374 5374 5374 33554432
if (kmerize:::check_install_ok()) { kmr_plot_response(res, ref_k = 9, max_y = Biostrings::width(dna)) }